Addressing Drug-Related SCARs
While rare, severe cutaneous adverse reactions to certain medications are potentially life-threatening and thus require immediate attention.
This course was published in the November 2019 issue and expires November 2022. The authors have no commercial conflicts of interest to disclose. This 2 credit hour self-study activity is electronically mediated.
EDUCATIONAL OBJECTIVES
After reading this course, the participant should be able to:
- Explain the role of oral health professionals in recognizing and managing the signs and symptoms of severe cutaneous adverse reactions (SCARs) to drugs.
- Describe clinical approaches to diagnosis and treatment for various SCAR conditions, as well as mortality rates for specific conditions.
- Discuss onset timing and the role of racial predilection for SCARs.
Severe cutaneous adverse reactions (SCARs) to drugs occur rarely, but can be life-threatening. Approximately 3% to 7% of all hospital admissions or referrals from primary care physicians are made due to adverse drug reactions of the skin.1,2 SCARs to drugs mainly consist of Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and acute generalized exanthematous pustulosis (AGEP). Each is categorized as a delayed-type hypersensitivity reaction, although the immune mechanisms differ between the presentations.
The incidence of SJS and TEN is estimated at 2 in 1 million people, whereas the incidence of DRESS syndrome is estimated to be 1 in 1,000 to 10,000.3 The estimated incidence of AGEP is one case to five cases per 1 million people per year, but may be underreported.4 While these reactions may be rare, dental practitioners must be aware of these potentially life-threatening symptoms early in their occurrence, and refer patients to a dermatologist for diagnosis and management (as most of these conditions are skin related). Early recognition and diagnosis of SCARs are crucial for minimizing adverse reactions and reducing morbidity.3 This article will review SCARs and present a case report of DRESS syndrome to illustrate diagnosis and management in a dental patient.
STEVENS-JOHNSON SYNDROME AND TOXIC EPIDERMAL NECROLYSIS
Variants of epidermal necrolysis, SJS and TEN are life-threatening mucocutaneous reactions that are similar in presentation and course, but differ in the extent of their involvement.3,5 With an onset of 4 days to 28 days after exposure, the initial manifestation is a prodromal fever and malaise, followed by a general physical deterioration and eventual dermatologic involvement.3 Flu-like symptoms, ocular involvement and skin pain often precede the mucocutaneous reaction and are important for early diagnosis. Initially, irregularly shaped erythematous macules and atypical target-like lesions present on the face, upper trunk, and proximal extremities, with progressive mucosal involvement and skin detachment.3,6 The erythematous lesions present a positive Nikolsky sign (clinical finding in the skin or oral mucosa in which the top layers of the skin or oral epithelium slip away from the lower layers when rubbed with a blunt instrument or finger), and blistering of the oropharynx, nasopharynx, eyes, genitalia or anus, which can lead to severe, debilitating, long-term sequelae or death.3
While primarily medication-associated, SJS and TEN have been reported to be caused by mycoplasm and viral infections, and also noted in patients with collagen vascular diseases.6 The most common drugs associated with this delayed-type hypersensitivity include sulfonamides, anticonvulsants, allopurinol, and nonsteroidal anti-inflammatory drugs (NSAIDs), but others have been documented as well.5 A genetic predisposition in different ethnic populations is seen, with particular HLA alleles activating a significant and specific immune response when exposed to certain medications.6 One such example is a 100% association between the HLA-B*15:02 allele and carbamazepine-triggered SJS/TEN in several Asian ethnicities.3 In short, the evidence suggests SJS/TEN appear to have a racial predilection, with a stronger association in individuals of African or Asian descent.6
The main pathophysiology involves CD8+ cytotoxic T lymphocytes, although NK cells and other antigen presenting cells have been implicated as well.1,3 A hallmark of these conditions is necroptosis, an apoptotic process that leads to the destruction of keratinocytes in SJS/TEN.1 For diagnosis, at least two mucosal areas must be affected, and the designation of SJS or TEN is based on the percentage of epidermis affected: SJS if less 10% of the skin is affected; TEN if more than 30% is affected; and an SJS/TEN overlap is designated if between 10% and 30% of the skin is affected.1,3 Treatment involves identification of the causative medication (via patient history or patch-test), and discontinuation of the drug. Additional treatment is palliative and symptom specific; SJS/TEN has a mortality rate between 10% to 40%, which varies depending on ethnicity.5 A summary of the main clinical and histological characteristics of SCARs is presented in Table 1.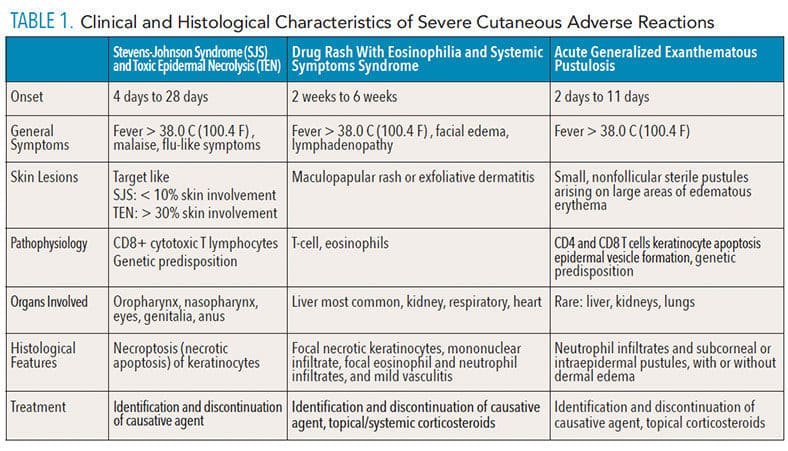
DRUG REACTION WITH EOSINOPHILIA AND SYSTEMIC SYMPTOMS SYNDROME
Also known as drug-induced hypersensitivity syndrome, DRESS syndrome is a delayed-type hypersensitivity reaction that is prone to misdiagnosis, and in approximately 10% of cases may prove fatal.6–8 Features include diffuse cutaneous involvement (maculopapular rash, or exfoliative dermatitis), fever, eosinophilia > 700 cells per µl, facial edema, lymphadenopathy, lymphocytosis, and systemic involvement, especially with the liver.1,6,8 Mucositis has also been identified in approximately 30% of patients.9 Early involvement might also include tachycardia and tachypnea.8
Most DRESS syndrome cases present 2 weeks to 6 weeks after initial drug exposure, although onset can occur as late as 16 weeks after exposure.6,10 Although causative agents include medications not often prescribed in dentistry (such as allopurinol or anticonvulsants), NSAIDs and certain antibiotics (such as ciprofloxacin combined with metronidazole and trimethoprim-sulfamethoxazole) have also been found to trigger DRESS syndrome.9 Importantly, ingestion of other medications, especially antibiotics, may promote DRESS syndrome and hasten the onset in patients whose disease course is due to other medications.6,10 ß-lactam antibiotics have been specifically shown to be related to the onset of DRESS syndrome, both alone or in combination with other medications.8
As symptoms progress, damage to organ systems begins to occur. The hepatic system is most commonly affected, followed by the renal, respiratory, and, perhaps most consequentially, the cardiac system.6 Cardiac involvement, although least reported, is the most likely to be associated with mortality.
The pathophysiology of DRESS appears to be multifactorial, with a T-cell-mediated immune response involving eosinophils being a hallmark of this disease. Once T-cells are activated, they release a variety of cytokines and chemokines that ultimately lead to the cutaneous presentation.6 Similar to that seen with SJS/TEN, a genetic predisposition involving multiple HLA alleles in patients of various ethnicities has been associated with DRESS syndrome.1,6,11 This is medication-specific, which supports the theory that individual HLA alleles would have an increased affinity for particular drugs, and, once exposed, will activate the adverse immune response described here.6,11 Many patients with DRESS syndrome also show serologic levels of particular viruses—mainly human herpesvirus (HHV)-6, HHV-7, Epstein-Barr virus, or cytomegalovirus.8–10 This reactivation of existing viruses has been suggested as the rationale for the delay in onset, progressive course, and systemic involvement that is seen with DRESS syndrome. Histologically, there is an eczematous pattern, with focal necrotic keratinocytes, mononuclear infiltrate, focal eosinophil and neutrophil infiltrates, and mild vasculitis.3 Diagnosis of the condition is often difficult, as multiple diagnostic criteria exist and the clinical presentation varies widely.3,10
In addition to identification and discontinuation of the causative medication, treatment of DRESS syndrome is usually with systemic and/or topical corticosteroids, although this can lead to unwanted immune suppression and viral reactivation, or other adverse side effects.3 An alternative treatment with cyclosporine has been indicated for cases in which prolonged steroid therapy is contraindicated or where systemic corticosteroids fail to work.7
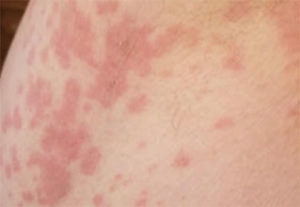
after the patient took amoxicillin, which the
dermatologist determined caused the condition.
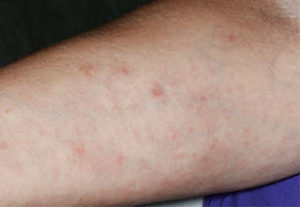
lesions on the patient’s right arm as they
presented 8 weeks after first appearing.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
Although AGEP has an onset of 2 days to 11 days after drug exposure, it typically occurs within 48 hours. Symptoms include high fever (> 38.0 C or 100.4 F), with simultaneous appearance of small, nonfollicular sterile pustules arising on large areas of edematous erythema.3 It is most commonly associated with these drugs: aminopenicillins, quinolones, hydroxychloroquine, sulfonamides, terbinafine, diltiazem, ketoconazole, and fluconazole.12 The areas that are first involved include the trunk and upper extremities (armpits and groin) and, during the initial stages, a positive Nikolsky sign may be present.
In less than 20% of AGEP cases, pustules or erosions develop on mucous membranes, usually of the oral cavity.12,13 Blood tests reveal elevated neutrophil counts (7,000 cells per ul), and mild eosinophilia.3,13 While AGEP rarely involves major organs (such as the liver, kidneys, or lungs), systematic investigations are highly recommended, as multiple organ involvement may require treatment in an intensive care unit.3,12 Histologically, most AGEP lesions show neutrophil infiltrates and subcorneal or intraepidermal pustules, with or without dermal edema.3 The main differential diagnosis for AGEP lesions are pustular psoriasis and bacterial folliculitis.3,12 In terms of histology, AGEP lesions appear with larger eosinophil infiltrates, more necrotic keratinocytes, and larger mixed dermal and interstitial infiltrates than are seen in pustular psoriasis; there is also an absence of dilated blood vessels.14 Additionally, there is often a personal or family history of psoriasis that may help differentiate between the two conditions.
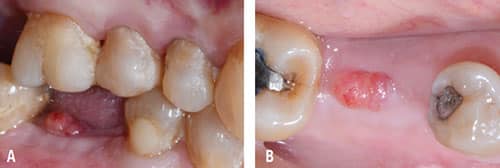
was part of the patient’s history, which is why it is included.
Pathophysiologically, exposure to the causative agent activates specific CD4 and CD8 T cells which, in turn, proliferate and migrate into the dermis and epidermis, causing keratinocyte apoptosis and epidermal vesicle formation.12 Certain genetic mutations may predispose individuals to develop AGEP.
As opposed to patients without a specific gene mutation, individuals with AGEP and the IL36RN gene mutation are more likely to have lip and oral involvement, and are considered predisposed to developing AGEP.12 This finding emphasizes the importance of a detailed review of the medical history and past history of reactions to medications. Diagnosis depends on clinical and histologic criteria. A drug patch test may be necessary to identify the cause of AGEP if the responsible drug is not clear.1 The main treatment is discontinuing the causative drug, which leads to significant symptom improvement within several days. During the pustular phase, moist dressings and antiseptic solutions may be used to prevent infection. Antibiotics should be avoided unless a superinfection of the pustules occurs. If pruritus and inflammation are present (as in prolonged cases), topical corticosteroids may also be appropriate.12 Finally, while use of systemic corticosteroids may provide relief, they may not reduce disease duration.4,12
CASE REPORT
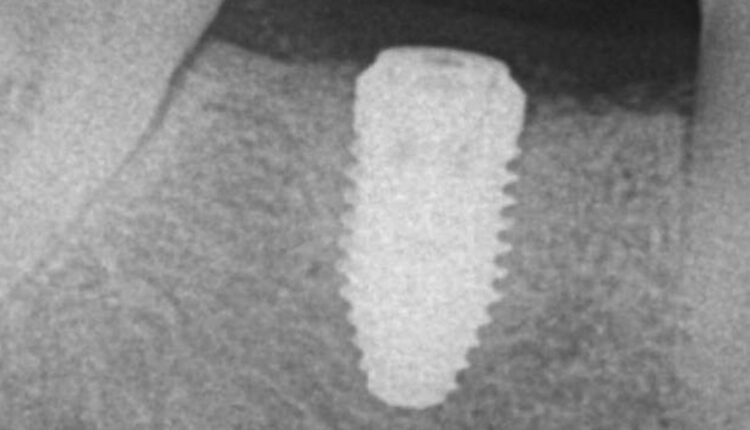
A 57-year-old Caucasian woman presented to the Louisiana State University Periodontics Department complaining of swelling on the mandibular right side of her jaw. At the time, the patient’s medical history included the following medications: pravastatin, omeprazole, and lorazepam. Endodontic evaluation determined tooth #30 was fractured. The patient elected to have the tooth extracted, with a ridge preservation procedure prior to implant placement. The patient was prescribed a 7-day course of 500 mg amoxicillin to be taken three times a day, along with ibuprofen 600 mg three times a day and hydrocodone/acetaminophen 5/325 mg for pain. The patient reported having no complications after taking these medications. Implant placement was completed 12 months later, after which the patient was again given the same regimen of antibiotics in addition to a methylprednisolone 4 mg dose pack (Figure 1).
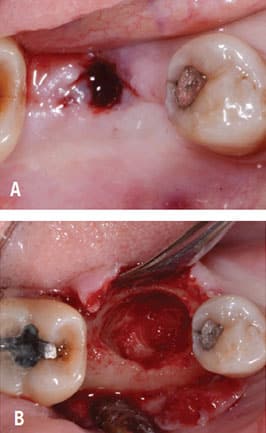
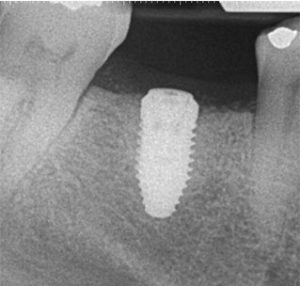
the site after implant was removed and the
implant site debrided.
Six weeks following implant placement, the patient began experiencing a widespread skin rash and pruritus (Figure 2). At this time, she was seen by a dermatologist to assess possible causes for the rash. After medical examination and skin patch testing, it was suspected the rash was a result of the amoxicillin that had been prescribed following implant placement. The dermatologist diagnosed the patient with DRESS syndrome, and she was prescribed a 14-day course of 10 mg prednisone. Her symptoms improved initially, but the rash returned 7 days after cessation of prednisone. At that point, she started an additional 20-day course of prednisone at 20 mg per day, which was tapered down to 10 mg for one week, and, finally, 5 mg for a final week. The patient underwent thorough allergy testing to rule out any additional causative agents, including titanium, and no positive reactions were noted. The rash gradually resolved within 8 weeks (Figure 3).
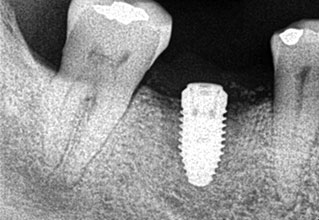
Following implant placement, the patient presented at 2 months with 5×5-mm granuloma at implant site #30 (Figure 4A and Figure 4B). Radiographs revealed extensive bone loss extending greater than 50% of the implant length (Figure 5). After consulting with the patient’s dermatologist to discuss pre- and postoperative management, the dental implant was removed, and, due to extensive bone loss, was able to be explanted with a hemostat (Figure 6A). After flap reflection and subsequent degranulation (Figure 6B), another grafting procedure was completed. At the direction of the dermatologist, the patient was prescribed doxycycline 100 mg and tramadol 50 mg for pain for 1 week. No complications with healing were noted after this procedure. This was an early implant failure of unknown etiology; however, it was not attributed to the DRESS syndrome. It is more likely due to the patient’s poor oral hygiene or lack of compliance. The future treatment plan includes a second attempt for implant placement in the area of #30. Following a consultation with the patient’s dermatologist, doxycycline 100 mg will be prescribed for 1 week if antibiotics are needed for that procedure.
CONCLUSION
This short review and case presentation highlight the necessity of early and accurate SCAR diagnosis. Although rare, these allergic reactions can be fatal. Thus, it is important for oral health professionals to be aware of the potential role of high-risk medications in triggering SCARs, and refer these patients in a timely manner to a dermatologist for identification of—and withdrawal from—the triggering medication. Immunologically, all SCARs discussed in this review present with CD8+ cytotoxic T Cell involvement, and, as noted, certain genetic mutations can predispose patients to developing SCARs.
REFERENCES
- Balakirski G, Merk HF. Cutaneous allergic drug reactions: update on pathophysiology, diagnostic procedures and differential diagnosic. Cutan Ocul Toxicol. 2017;36:307–316.
- Doña I, Barrionuevo E, Blanca-Lopez N, et al. Trends in hypersensitivity drug reactions: more drugs, more response patterns, more heterogeneity. J Investig Allergol Clin Immunol. 2014;24:143–153; quiz 1 p following 153.
- Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390:1996–2011.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:pii:E1214.
- Miliszewski MA, Kirchhof MG, Sikora S, Papp A, Dutz JP. Stevens-Johnson syndrome and toxic epidermal necrolysis: an analysis of triggers and implications for improving prevention. Am J Med. 2016;129:1221–1225.
- Chen CB, Abe R, Pan RY, et al. An updated review of the molecular mechanisms in drug hypersensitivity. J Immunol Res. 2018;2018:6431694.
- Kuschel SL, Reedy MS. Cyclosporine treatment of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome: a case report and brief review of the literature. Pract Dermatol. 2018;2018:41–43.
- Girelli F, Bernardi S, Gardelli L, et al. A new case of DRESS Syndrome induced by sulfasalazine and triggered by amoxicillin. Case Rep Rheumatol. 2013;2013:409152.
- Ang CC, Wang YS, Yoosuff EL, Tay YK. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Derrmatol. 2010;63:219–227.
- Corneli HM. DRESS Syndrome: drug reaction with eosinophilia and systemic symptoms. Pediatr Emerg Care. 2017;33:499–502.
- Phillips EJ, Bigliardi P, Bircher AJ, et al. Controversies in drug allergy: testing for delayed reactions. J Allergy Clin Immunol. 2019;143:66–73.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843–848.
- Sidoroff A, Halevy S, Bavinck JN, Vaillant L, Roujeau JC. Acute generalized exanthematous pustulosis (AGEP) — a clinical reaction pattern. J Cutan Pathol. 2001;28:113–139.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: Does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609–611.
From Dimensions of Dental Hygiene. November 2019;17(10):30—33.
