 LIGHTHAUNTER / ISTOCK / THINKSTOCK
LIGHTHAUNTER / ISTOCK / THINKSTOCK
Caring for Patients With Huntington’s Disease
This genetic disease has severe consequences for oral and overall health.
This course was published in the September 2016 issue and expires September 2019. The authors have no commercial conflicts of interest to disclose. This 2 credit hour self-study activity is electronically mediated.
EDUCATIONAL OBJECTIVES
After reading this course, the participant should be able to:
- Define the etiology of Huntington’s disease.
- Discuss signs and symptoms of each stage of Huntington’s disease.
- Address how this genetic condition is managed and treated.
- Identify modifications needed to provide dental hygiene care for patients with Huntington’s disease.
The early symptoms of Huntington’s disease include chorea (presence of involuntary jerky movements) and loss of balance, but individuals may show cognitive or psychiatric impairments years before reduced motor symptoms start to appear.2 The mean age of onset for symptoms is 40, and both sexes are equally affected.3 The average lifespan after the onset of Huntington’s disease is 10 years to 20 years.4 Death is usually caused by secondary conditions, such as respiratory infections, physical injuries related to falls, or complications with swallowing and/or aspirations.5 The suicide rate among individuals inheriting Huntington’s disease is five times greater than the general population, as many of these individuals have witnessed a family member become emaciated and immobile.5
ETIOLOGY
A gene is a sequence of nucleotides that code in order to produce a certain protein. There are four main nucleotides: adenine (A), cytosine (C), guanine (G), and thymine (T). The defective gene that causes Huntington’s disease was discovered in 1993 and is located on chromosome four.6 Ordinarily, this gene makes a protein called huntingtin (HTT), which is essential for normal neural development and is used primarily in the cerebral hemisphere and central nervous system.7 Huntington’s disease results from a mutation in this gene that causes an abnormal repetition of cytosine, adenine, and guanine (CAG) sequence of nucleotides.3 Forty or more of the CAG repeat sequences create a protein that misfolds and becomes unfunctional, leading to Huntington’s disease.
Four classifications are used to define individuals’ risk of developing Huntington’s disease based on their CAG repeat. The first classification is defined as normal, which means they have a sequence of CAG repeats of 26 or less; these individuals have no risk of acquiring the disease or passing it to their offspring.6,8 The second classification is intermediate. These individuals have 27 to 35 CAG repeats. They have no risk for developing the disease and they have a less than 50% risk of passing the mutation to their offspring.6,8 The third classification is reduced penetrance, and these individuals have a CAG repeat count of 36 to 39. Individuals in this category may or may not develop the disease, and there is a 50% chance of passing the mutation to their offspring.6,8 The fourth classification is entitled full penetrance and these individuals have a CAG repeat sequence of 40 or more and will develop Huntington’s disease. Full penetrance classification individuals have a 50% chance of passing the mutated gene to their offspring.6,8 The more CAG repeats the person has, the earlier the onset of the disease or increased severity of the disease.6
The mutation in the HTT gene produces a mutant huntingtin protein (mHtt), which results in an extended number of CAG repeats. The normal role of huntingtin protein (Htt) is not clearly known, but it does seem to be essential in normal neural development and nerve cell function.4 HHT regularly interacts with proteins found only in the brain. Thus, altered HHT is most disruptive to nerve cells.9Researchers have theorized that the mHtt can lead to cell damage and toxicity through protein aggregation and/or neuronal inclusions.10 The process of protein aggregation causes the protein to misfold or clump, which can interfere with normal cell function. In the altered HHT protein, the extended number of glutamine repeats causes links to form within and between proteins.9 Rather than folding into functional proteins, they tangle up into protein aggregates—which can disrupt the cell’s nucleus from functions such as transcription or mitochondrial activity, impairing the energy production in a cell.9,11
Another theory relates to neuronal inclusions or inclusion bodies, which are also produced by the excessive glutamine in the mHtt.11The neuronal inclusions initially form in the axons and dendrites in the neural cells, blocking axonal transport from occurring in the cell.11 Dysfunctional axonal transport causes the distal axon to degenerate, inhibiting nerve impulses or synapses from taking place and halting communication.12 As the neuron cell dies, macrophages clear out the decayed debris.13 More research is needed to establish the exact pathophysiology.
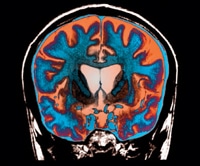
STAGES OF DISEASE
While the signs and symptoms of Huntington’s disease may vary among individuals, the progression of the disease can be divided into three stages: early, middle, and late. Cognitive or psychiatric impairments may appear many years before reduced motor symptoms start to appear.2 The date of diagnosis is not a reliable indicator of the stage of disease, as the progression of symptoms does not occur in a predictable order.14 Each stage of the disease lasts approximately 5 years and spans over 15 years to 20 years of average life expectancy at the onset of symptoms.7 The clinical manifestations of Huntington’s disease typically begin in midlife with characteristic motor abnormalities, such as uncoordinated movements (chorea), loss of muscle control (dystonia), swallowing complications (dysphagia), personality changes (depression, irritability), a gradual loss of cognition, and, eventually, death.15
During the early stages of Huntington’s disease, the basal ganglia—the part of the brain that controls behavior and movement—is impacted the most seriously (Figure 1). Early symptoms usually include minor changes in coordination and difficulty with organizing, prioritizing, or focusing on tasks (Table 1).1,5 The individual may experience depression and irritability.1 Most individuals present with neurological and psychiatric symptoms that can affect their ability to work and accomplish self-care.14 Although death is rare at this stage, thoughts of suicide are common.7 In addition, acceptance of the disease and its inevitable outcome causes uncertainty for the patient’s family.
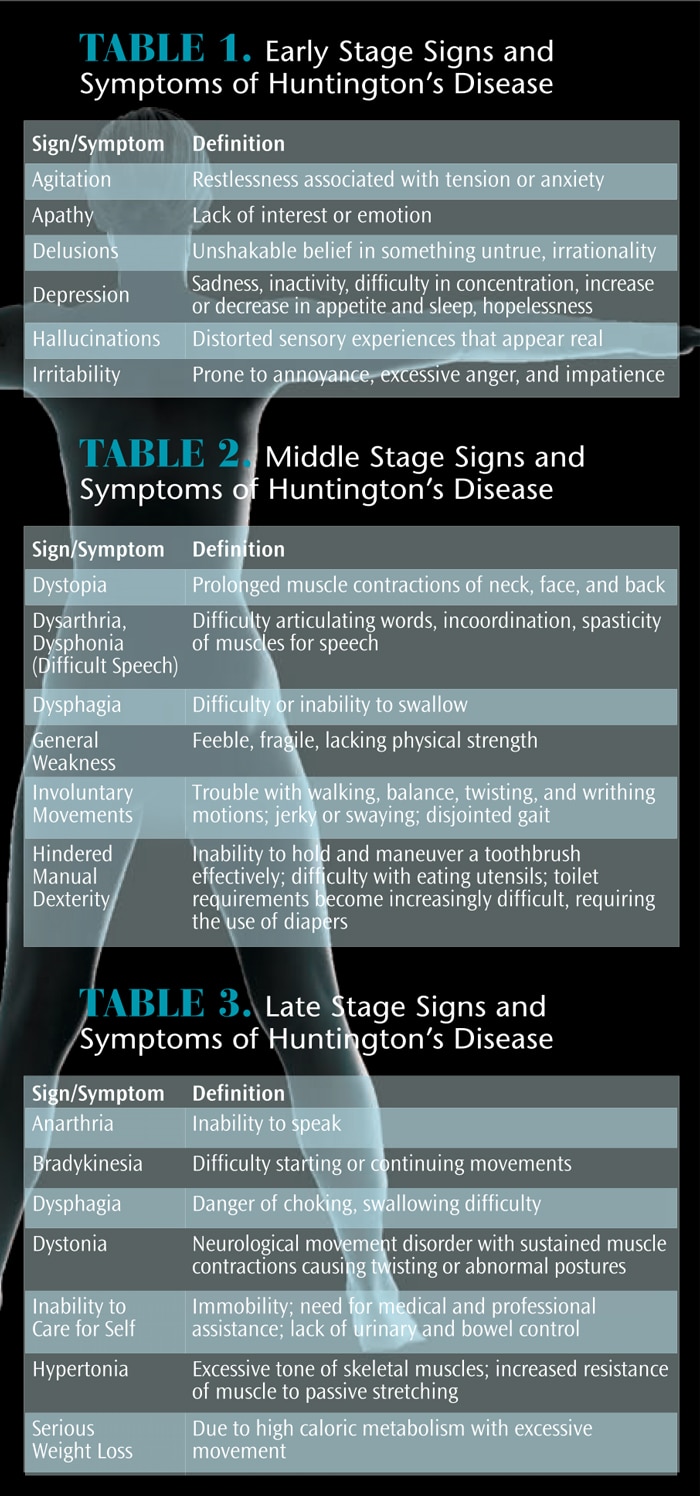
As Huntington’s disease advances into the middle stage, involuntary movements become more obvious (Table 2).14 Speech and swallowing are increasingly difficult and ordinary activities become more challenging.1 Although most individuals in the middle stage can feed themselves, they may become more dependent on caregivers.14 Individuals are usually unable to remain employed due to a decline in cognitive ability.14
In the late stages of Huntington’s disease, the individual is no longer able to live independently and self-care declines or is nonexistent (Table 3). Cognitive deterioration is obvious and swallowing difficulties and involuntary movements make it impossible for individuals to feed themselves. Performing daily oral hygiene becomes impossible without assistance.14 Most can no longer walk or speak.1 At this point, most patients are transitioned into a long-term care facility, requiring around-the-clock care.7
MANAGEMENT/TREATMENT
Currently, there is no cure for Huntington’s disease, nor are treatments available to halt or slow disease progression. Treatment is aimed at relieving and managing symptoms. This may involve emotional support as the patient expresses behavioral and physical difficulties. Due to the complexity of the disease, an interprofessional approach requiring the assessment and interventional treatment by a multidisciplinary team is the most prudent approach.14 Pharmacological and nonpharmacological therapies are available for patients with Huntington’s disease. A patient-centered approach should address personal hygiene; nutrition and hydration; communication strategies; health, safety, and mobility; and swallowing activity.7 Caregivers of patients with Huntington’s disease should be involved along with primary care physicians, neurologists, psychiatrists, dietitians, occupational therapists, social workers, speech and language therapists, physical therapists, and oral health professionals.14
A review of the current literature establishes the importance of preventive dental care and treatment for patients with Huntington’s disease.12 The primary goal of dental treatment planning should be the preservation of the natural dentition for as long as possible. Pathologic conditions can be diagnosed and treated as the disease progresses. Eventually, the patient’s choreiform movements may become too difficult to control. The use of oral sedatives or intravenous (IV) sedation may enable the provision of preventive and restorative care in a more effective and safe manner.12 Many patients with Huntington’s disease start to experience anxiety in the more advanced stages, and sedation can aid with reducing some of their apprehensions (see case study).
Movement and psychiatric disorders can be managed with oral medications, such as tetrabenazine and clonazepam, to control chorea and involuntary movements.5 Anti-psychotic drugs like chlorpromazine, haloperidol, quetiapine, and risperidone are used to control the hallucinations and violent outbursts.5 Anti-depressant medications, such as citalopram, fluoxetine, and sertraline, are prescribed to address depression that many individuals experience.5 All of these medications can cause xerostomia, and some may also lead to gingival enlargement and/or glossitis.14
Caregivers should be informed of the need for frequent preventive dental visits, which may be every few weeks or every few months, depending on the individual’s needs. It is also important to focus on meticulous daily oral cleaning. This is necessary whether the patient has a partial dentition, full dentures, or a combination of implants and natural teeth. As the disease progresses, motor function declines, salivary flow decreases, and the incidence of periodontal disease severity and caries risk increases.12 In order to provide continuity of care and create familiarity, care should be provided by the same clinician at each visit when possible.
MODIFICATIONS TO CARE
Caring for patients with Huntington’s disease can be challenging, especially as the disease progresses into the middle and late stages. Due to patients’ inability to perform adequate self-care, a significant amount of biofilm, food debris, and calculus is often present. As the ability to chew and swallow diminishes, a soft diet is introduced, which may raise the risk of dental caries. Caregivers may also introduce high caloric foods and cariogenic drinks or snacks to maintain energy, which is depleted by the spastic, uncontrolled movements.14
Dental hygienists need to work collaboratively with caregivers and the interprofessional team. To obtain optimal oral health, frequent recare appointments are required. Depending on the patient’s preference, morning appointments are recommended as weakness and fatigue increase throughout the day.16 Additional assistance for the appointment varies depending on the patient’s ability to walk. Safe wheelchair transfer techniques need to be implemented if the patient is confined to a wheelchair. In the middle or late stages of the disease, the patient may experience difficulty swallowing and the clinician will have to adapt care accordingly by seating the patient in a more upright position, using ideal suctioning, and limiting devices requiring excessive water and cavitation.16 Bite blocks may assist patients with muscle weakness and limited ability to keep their mouths open.16 In addition, the caregiver can help stabilize the patient’s head or hold the patient’s hand for assurance and to reduce anxiety. As the disease progresses into the middle and late stages, the use of IV conscious sedation or general anesthesia may be needed in order to provide dental and dental hygiene care.14
Oral self-care education for patients with Huntington’s disease depends on many factors, such as motor/dexterity abilities and cognitive ability. In the early stage of the disease, patients may be able to brush with a power toothbrush, until the weight of the toothbrush becomes too heavy. Other options include modifying manual toothbrush handles to enlarge the diameter with a bicycle handle, a tennis ball, or even an empty soda can.16 Traditional flossing becomes too challenging as the disease progresses. The handle of an extended flosser can be modified, as well, to enable the patient to grip the handle better.16 Other interproximal cleaning devices such as oral irrigators could pose concern for the patient who is experiencing a decline in swallowing as the disease advances. Mouthrinses should also be avoided in the later stages to circumvent aspiration.14 Use of salivary substitutes and xylitol and/or fluoride products can aid in the reduction of xerostomia and caries prevention.17 Throughout the disease process, caregivers should be encouraged to provide oral health care daily as the abilities of the individual with Huntington’s disease decline. Oral health professionals need to assess the caregiver’s oral health status, as well. Research has shown caregivers who value their own oral health provide better care to the individual who requires their assistance.18
CASE STUDY
A 48-year-old man presented to the dental office with his older brother, who is his caregiver. The patient’s chief compliant was “broken teeth” that caused him pain when he chewed. He had been diagnosed with Huntington’s disease 6 years prior. His brother reported that the patient was in the middle stage of the disease.19
The patient exhibited choreiform movements, occurring randomly several times per minute and disturbance of gait. The patient was able to remain still for periapical radiographs, but expressed apprehension about needing to sit still during dental treatment for several hours at a time. The patient’s brother also noted that the patient has significant dental anxiety, along with anger outbursts if he feels he has no control of a situation.19
Due to the extensive treatment plan requiring both nonsurgical periodontal therapy and restorative care, IV sedation was deemed necessary. The patient consented to have all hopeless or marginal teeth extracted. The patient refused to have any type of removable or permanent prosthesis because of cost. The patient’s brother had concerns regarding removable prosthesis due to the patient’s choreiform movements, which would only exacerbate as his disease progresses. Fortunately, a sufficient number of occluding teeth remained after treatment to maintain the ability to chew.19
Quadrant periodontal debridement and restorative care were performed over four separate appointments with IV sedation. The patient tolerated the treatment well. At 1-week post-operative, the extraction sites were healing well and the patient reported he could chew without any pain and experienced fewer swallowing problems. The patient was placed on a 3-month recare program for periodontal maintenance. Although the patient has choreiform movements, he should be able to tolerate recare appointments with frequent breaks, handholding by a caregiver, and head stabilization. The patient currently uses a power toothbrush to control biofilm, which is semi-adequate at this phase of the disease. The patient’s brother has taken over the interdental cleaning aspect of self-care as the patient can no longer perform these tasks. The self-care practices will need to be modified as the patient’s ability to perform daily living activities declines.19
CONCLUSION
Dental hygienists can be an important resource for individuals with Huntington’s disease and their caregivers by providing education and care through all stages of the disease. Dental hygienists work collaboratively with the interprofessional team as the disease affects the individual’s abilities and requires a diverse group of specialized health care professionals to ensure all aspects of the patient’s health care needs are met.13 As devastating as this disease can be, dental hygienists can provide the preventive oral care needed to ensure patients with Huntington’s disease can maintain optimal oral health for as long as possible.
References
- Huntington’s Disease Society of America. What is Huntington’s Disease? Available at: hdsa.org/what-is-hd/?gclid=CL_Y7Jvgvs0CFZJlfgodrvsD_w. Accessed August 21, 2016.
- Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79: 874–880.
- Gonzalez-Usigli HA. Merek manual professional version- Huntington’s disease. Available at: merckmanuals.com/professional/neurologic_disorders/movement_and_cerebellar_disorders/huntington_disease.html. Accessed August 21, 2016.
- Novak MJ, Tabrizi SJ. Huntington’s disease. Brit Med J. 2010;341:34–40.
- Mayo Clinic. Huntington’s Disease. Available at: mayoclinic.org/diseases-conditions/huntingtons-disease/basics/definition/con-20030685. Accessed August 21, 2016.
- Hogarth P. Huntington’s disease: a decade beyond gene discovery. Curr Neurol Neurosci. 2003;3:279–284.
- Baker M. Huntington’s disease part 1: what is it? British Journal of Healthcare Assistants. 2009;3(5):223–227.
- Squitieri, F, Jankovic J. Huntington’s disease: how intermediate are intermediate repeat lengths? Mov Disord.2012;27:1714–1717.
- Liou S. HD Genetics. Available at: web.stanford. edu/group/hopes/cgi-bin/hopes_test/huntingtin-protein-and-protein-aggregation/#the-huntington-gene. Accessed August 21, 2016.
- Cisbani G, Cicchetti F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity.Cell Death Dis. 2012;3:e382.
- Chang D, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400.
- Saft C, Andrich JE, Muller T, Becker J, Jackowski J. Oral and dental health in Huntington’s disease- an observational study. BMC Neurol. 2013;13:114–118.
- National Institute of Neurological Disorders and Stroke. Available at: ninds.nih.gov/disorders/brain_ basics/ninds_neuron.htm. Accessed August 21, 2016.
- Manley G, Lane H, Carlsson A, et al. Guideline for oral health of adults with Huntington’s disease. Neurodegen Dis Manage. 2012;2:55–65.
- Auday BC, Buratovich MA. Magill’s Medical Guide. 7th ed. Hackensack, New Jersey: Salem Press; 2014;3:1148.
- Darby ML, Walsh MM. Dental Hygiene Theory and Practice. 4th ed. St. Louis: Elsevier Inc: 2015:872–890.
- Harris NO, Garcia-Godoy F, Nathe CN. Primary Preventive Dentistry. 8th ed. Upper Saddle River, New Jersey: Pearson Education Inc: 2013:53–68.
- Limeres J, Martinez F, Feijoo JF, Ramos I, Linares A, Diz P. A new indicator of oral hygiene habits of disabled persons: relevance of the carer’s personal appearance and interest in oral health. Int J Dent Hyg. 2014;12:121–126.
- Rada Robert E. Comprehensive dental treatment of a patient with Huntington’s disease: literature review and case report. Spec Care Dent. 2008;28:131–135.
From Dimensions of Dental Hygiene. September 2016;14(09):41–44.
